INTRODUCTION TO PROTEIN DESIGN RESULTS
Our Design tools are often used as a way to find multiple mutations that improve Rosetta Energy. We recommend running 100 repeats of a design. Then look at the range of results to find favorable mutations. Instructions on how to access and use the Sequence Logo do can be found here or here.
If you are not getting as much sequence variety as you expect, you can increase the number of repeats to 500 or more. If more variation does not occur, then you have either found the best sequences or your protein structure is not fully optimized.
Having more results can often be more informative, but they can become difficult to analyze. Here we describe how to find unique sequences in a set of design repeats. First, download the sequence of all design results. To do this begin by clicking on the folder with the design results in the left panel of Cyrus Bench to bring the design results into the center. Select all structures by clicking the box above the list of structures (shown as Step 1 below). Click ![]() and choose FASTA sequences (Step 2 below).
and choose FASTA sequences (Step 2 below).

FIND AND COUNT UNIQUE SEQUENCES
Use this workflow if you have access to MS Word and Excel (or similar programs) but lack software for alternative multiple sequence alignment.
1.) Preparing FASTA File with Design Results
Open the FASTA file which contains all the structures generated in your design results using MS Word or another program capable of “Find and Replace”. See the instructions above for how to download this FASTA file. In MS Word, click Edit > Find > Replace to open a window as shown below:
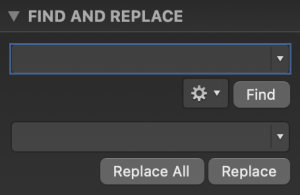
Remove all “end of line” indicators by entering ^p in the Find field and leaving the Replace field empty. This will make all the output in the same line.
Add an “end of line” between each sequence by entering > in the Find field and ^p in the Replace field.
If the first 4 residues of the FASTA sequence are not repeated within the sequence and were not included in the design process: Add a comma at the beginning of the sequence by entering the first 4 letters of the sequence in the Find field and a comma followed by the first 4 letters of the sequence in the Replace field empty as shown below:

Finally, remove sections of the sequence names that are not needed. For example enter relax_design 35 results | structure in the Find field and leaving the Replace field empty.

You may leave your FASTA sequences as they are. if you would prefer to cut down the sequence to the region(s) where mutations were allowed in the design run you can use Find/Replace as needed to delete regions of the sequence where you know no mutations were made.
Note: When using Find/Replace in Word, double check whether you have made a mistake by checking how many times Find/Replace made a replacement. If you have 100 sequences, then there should have been 100 replacements.
2.) Converting FASTA File with Design Results to an Excel Spreadsheet
Copy all the results of your sequences from Word as described in part one and paste into an Excel spreadsheet.

Select the cells containing your data in Excel, they should all be in one row as shown above. Click Data > Text to Columns. This should bring up a window like this:
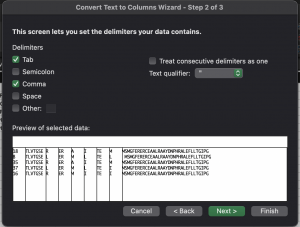
Choose Delimited, click Next. Select comma, click Next, then Finish.
Label the new columns for the data as follows:
- Type Structure # in a cell above the column containing the structure number
- Type Position or Pos_1, Pos_2, etc in a cell above the column with the structures’ sequences.
Select the cells that include your data then click Data > Sort. Click the region under Column and choose Sequence. Then click + in order to add another level of sorting. Click the region under Column for the next section and choose Structure # as shown below. Click OK.
In this example, there are multiple positions separated to allow sorting by multiple positions:
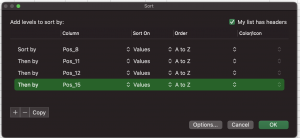
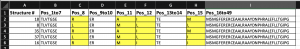
This will sort your data so that each unique sequence is clustered together and will list the lowest structure number first.
Add another column to the right of the existing data called Unique as shown below. In the first cell under the new column Unique, use an If/And formula to find unique sequences by typing the following =IF(AND(C1=C2,E1=E2,F1=F2,H1=H2), “Repeat”, “Yes”) as shown below:
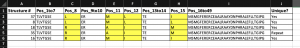
This IF/And formula will look at the sequence in the current row and compare it to the row above it for all 4 columns that contain mutating positions. If they are the same, the Unique column displays Repeat. If the sequences are not the same the Unique column for that sequence displays Yes. Copy and Paste Special the “value” in these cells for the entire column because we want to Sort. Sorting will change the order and thus change the value.
Sort the section by Unique? to separate the repeats from the unique sequences.