OVERVIEW
Homology Modeling (HM) is the most effective method for predicting protein structure from its sequence. This tool uses the structures of homologous proteins, or protein fragments, as an estimate for your protein structure and then models conformational differences that are likely to exist due to sequence divergence.
BACKGROUND
If you would like to learn more about how HM works you may find this step-by-step diagram and explanation helpful. This explanation will walk you through how HM aligns your sequence to homologs in the PDB, then undergoes 200 rounds of modeling for your sequence, before sorting through the results to present you the best structure predictions. If you would like a more technical in-depth description of how HM click here.
RUNNING HOMOLOGY MODELING
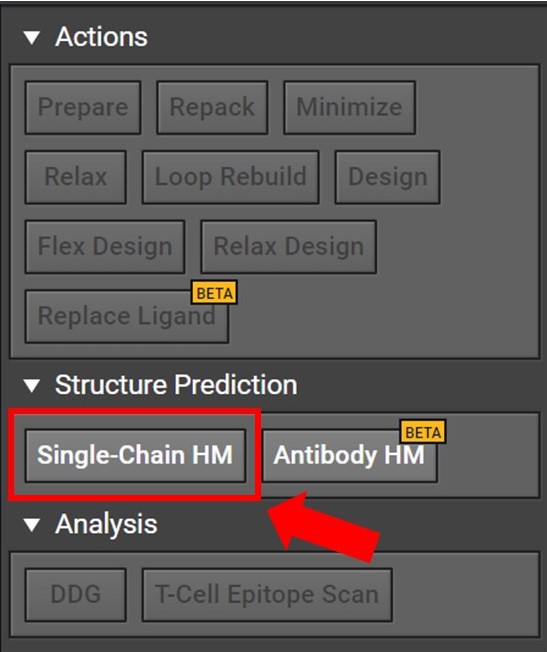
Begin by opening a project in CAD and select the Single-Chain HM button located to the right under Structure Prediction as shown below.
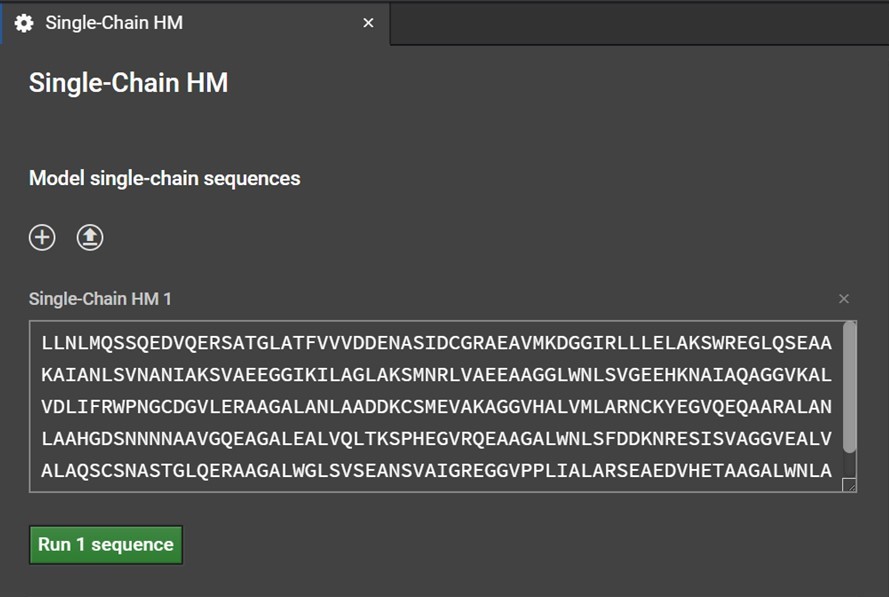
Clicking Single-Chain HM button will open a tab in the center window. Enter your sequence into the box as shown below. Please note, currently HM can only model the canonical 20 amino acids: do not enter non-canonical letters or symbols including spaces or else an error warning will be issued.
You can also click ![]() in order to upload a file containing FASTA format sequences of one or more runs. Clicking
in order to upload a file containing FASTA format sequences of one or more runs. Clicking ![]() will bring up another box if you would like to paste another sequence. Once you have entered or uploaded your sequences, click
will bring up another box if you would like to paste another sequence. Once you have entered or uploaded your sequences, click ![]() and your job will begin processing.
and your job will begin processing.
PLEASE NOTE: if you are learning to use HM you may choose not to run example sequences because all HM runs will be charged to your account. You may find it helpful to follow this beginner’s tutorial on using HM which will guide you through an example sequence and includes downloadable files for the final PDB structures generated, sequences, and alignments.
HM is the most time-consuming process that can be run; the length of the run time is dependent on the length of your sequence. If you have a 1,000 residue structure, it could take well over 24 hours to process. A 25 residue structure could be done in under an hour.
HOMOLOGY MODELING RESULTS
To open your HM results in CAD click on the ![]() results folder for your finished run, located in the left window. This will open a new tab as shown below.
results folder for your finished run, located in the left window. This will open a new tab as shown below.
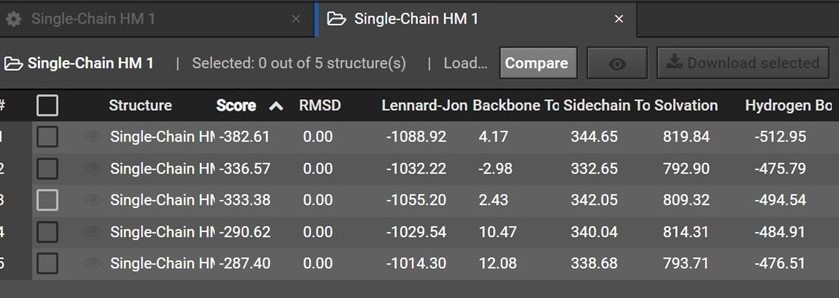
During HM 200 structures are predicted and are then grouped by shape into five clusters. Each of the five structures in your HM result is a representative structure from one of these clusters. The clusters and representative structures are ordered in your results by score.
Please note however that the second structure is not the second best structure, instead this structure should be used as an indication of how similar the first and second clusters are.
Assessing the Structure Prediction Results
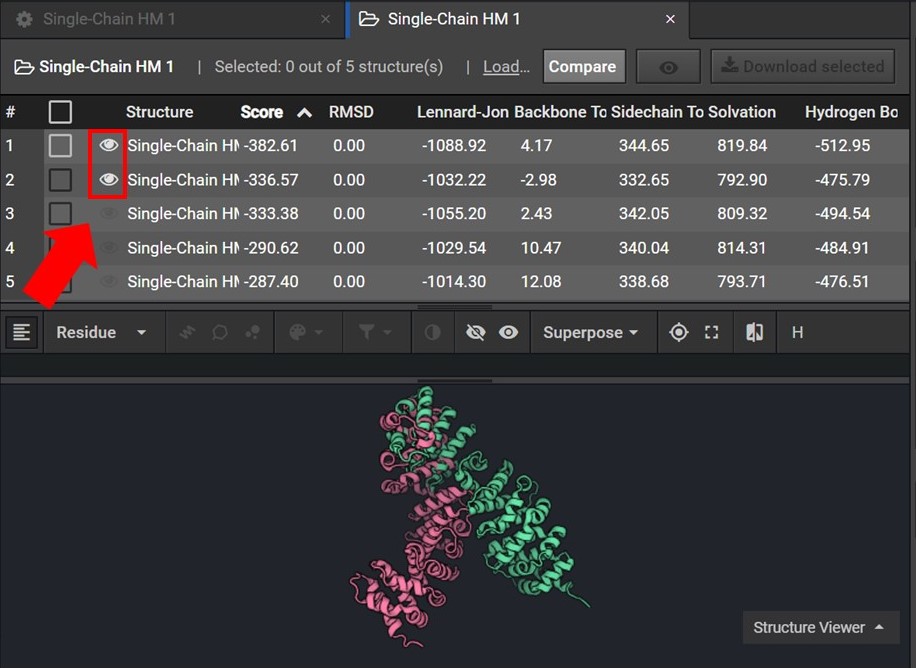
To assess the accuracy of your results first sort your results by Score and then view the two lowest score structures in the Structure Viewer by selecting ![]() next to each structure name as shown below.
next to each structure name as shown below.
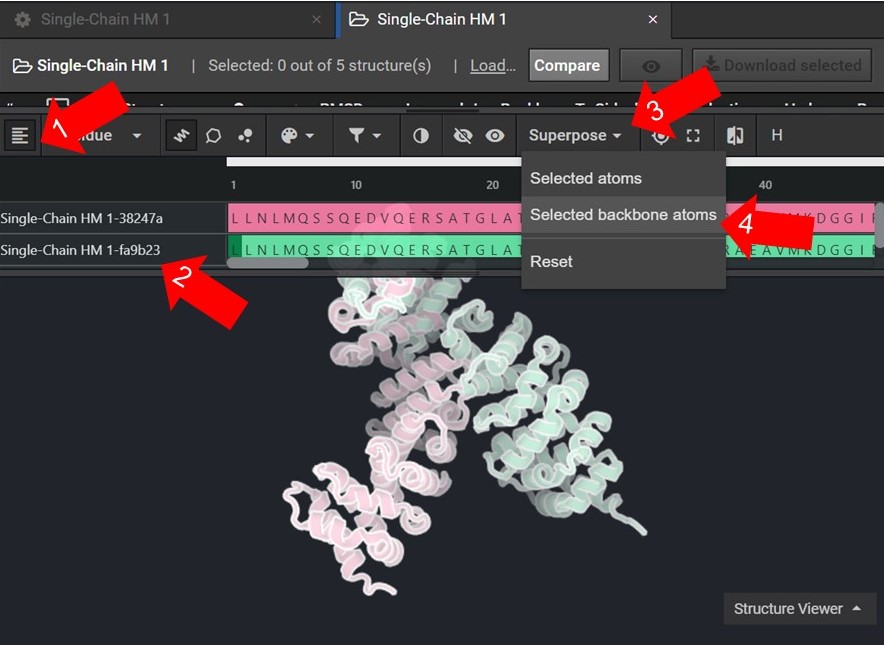
As shown below click ![]() to view the structures’ sequences (Arrow 1) and highlight both sequences by click on each sequences’ name(Arrow 2). Then use the superpose tool (Arrow 3) to align the two structures by backbone atoms (Arrow 4). For a more in-depth tutorial on these CAD features please visit Tutorial 1 and Tutorial 2.
to view the structures’ sequences (Arrow 1) and highlight both sequences by click on each sequences’ name(Arrow 2). Then use the superpose tool (Arrow 3) to align the two structures by backbone atoms (Arrow 4). For a more in-depth tutorial on these CAD features please visit Tutorial 1 and Tutorial 2.
If the two structures do not seem to converge after they have been superposed then your HM run may not have reached the energetically minimum structure. To improve the accuracy of your results select the two lowest energy structures from your HM run as input for 20 rounds of Relax. Then input the lowest energy structure from the Relax runs into another HM run.
Using and Accessing the Alignment File
With your HM structure predictions your results include additional information in an alignment file about which known structures were aligned with your sequence. You may find this information helpful when evaluating what regions of your predicted structure are most accurate, as well as illustrating where gaps were in the alignment.
Each alignment between a template structure and your sequence has a corresponding weight indicating the confidence in the alignment. The best alignment receives a weight of 0.6 and the weights decrease from there. The higher the weight of an alignment, the less the predicted structures will be allowed to deviate from the template during the modeling process.
As shown below the alignment file can be accessed from the HM run tab, if you have closed this tab click the run job title which is located to the right in a list under Structure Prediction.
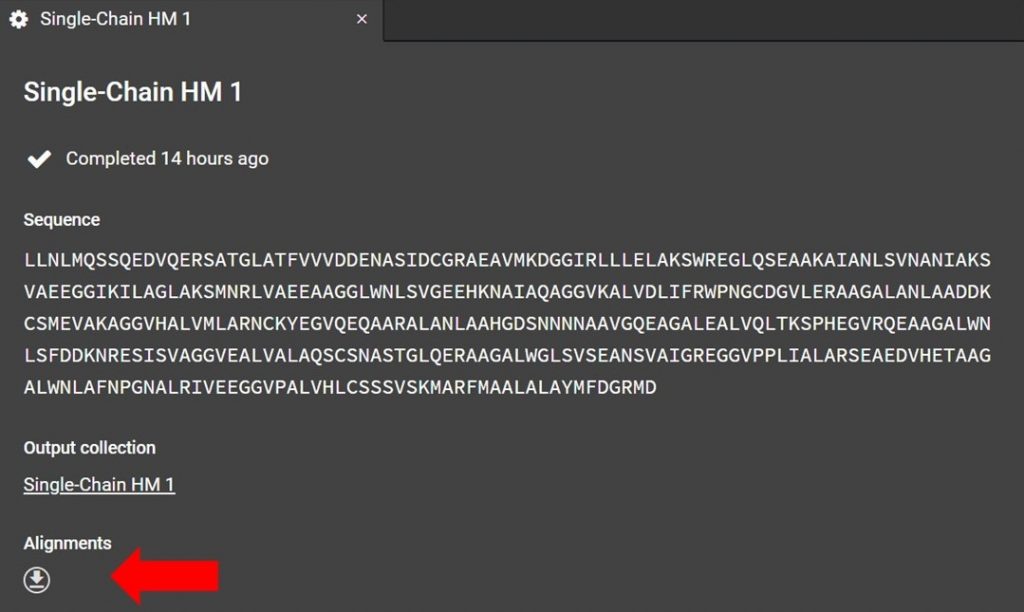
Click as indicated above to download the alignment file. To read the alignment file open it with any text editor that supports monospaced font, for example Atom.
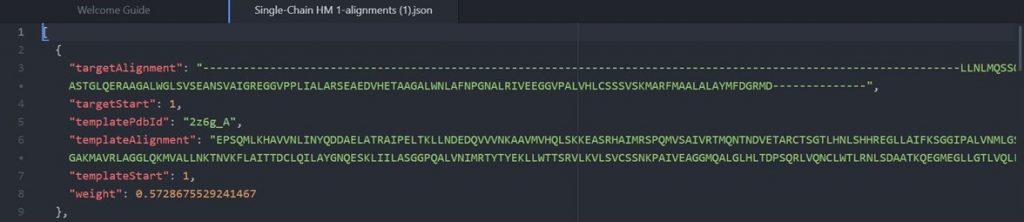
As shown above the alignment file will display the original target input sequence with each template sequence.
Each alignment can also be better visualized by entering the target and template sequences into Clustal Omega Multiple Sequence Alignment as shown below.
