Delta Delta G (DDG) is a metric for predicting how a single point mutation will affect protein stability. To read more background information on DDG click here.
1.) Begin in CAD by clicking the folder on the left that contains your protein structure to bring it into the center window.
2.) Click the box next to the structure so that a check mark appears in the box. Then you can click the ![]() button on the right lower panel under Metrics to bring the run into the center window. By default, the selector will say
button on the right lower panel under Metrics to bring the run into the center window. By default, the selector will say ![]() , but you can switch to one of the selectors that you have created. For more information on how to create and use selectors click here. The chart showing all original sequences and all potential point mutations is shown in the bottom center.
, but you can switch to one of the selectors that you have created. For more information on how to create and use selectors click here. The chart showing all original sequences and all potential point mutations is shown in the bottom center.
![]()

3.) You can select (or deselect) all mutations at all locations by clicking ![]() in the upper left hand corner. You can also click an entire row or column, as well as any individual box. Original sequences cannot be selected because the 𝚫G is 0.
in the upper left hand corner. You can also click an entire row or column, as well as any individual box. Original sequences cannot be selected because the 𝚫G is 0.
You can also upload your desired mutations by clicking. Once you have selected all the mutations that you want to measure, click ![]() . Before beginning the run your selections show as
. Before beginning the run your selections show as ![]() ,and will show as
,and will show as ![]() while the calculation is running. Once Design is done it will show the 𝚫G.
while the calculation is running. Once Design is done it will show the 𝚫G.

![]()
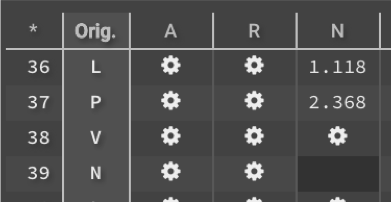
Negative 𝚫G indicates that the mutation will improve stability while positive 𝚫G will disrupt stability. You can download the entire chart by clicking ![]() .
.
Note: Running DDG for residues at a protein-ligand interface needs to be run differently, this additional tutorial may be helpful. In brief DDG may be run as follows for protein-ligand interfaces:
First, run DDG for the protein-ligand complex, then repeat the same calculation for the protein alone. This will separate the energetic changes that happen with the protein alone vs stabilization of the interface.
DDGinterface stability = DDGcomplex – DDGprotein
DDG has not been benchmarked for this specific use.
UNDERSTANDING AND USING DDG RESULTS
DDG results will fall into three categories:
1.) DDG > 0.5: Positive results suggest that a mutation would be destabilizing. Most mutations will be positive or close to zero because proteins have evolved to be reasonably stable. These mutations are residues that you should usually avoid during design.
2.) 0.5 > DDG > -0.5: 𝚫G that are near 0 are within the noise range so should be considered neutral. These can be included in design to allow more neutral changes in the protein that may compensate for changes in the protein made during design.
3.) DDG < -0.5: Negative results suggest that the mutation would lead to a more stable protein. However, the environment at each position should be considered.
- If interacting molecules are not present in the model, such as at a known zinc binding site, then a seemingly favorable mutation will not be favorable in reality.
- A position that has a a very negative DDG could mean that this position evolved a destabilizing residue because it is necessary for its catalytic activity, for binding another molecule, or because of another functionally relevant reason.
- Also, consider that this measures a single point mutation. Many times it requires multiple interacting mutations in order to achieve significant stability.
DDG is only predictive for a single mutation at a time. In order to predict whether multiple mutations would provide an additive effect on stability, you must create the mutations in design and run some Relax repeats. Then the mutated structures energy must be compared to the original structure (which also needs Relax repeats to be comparable). This is nearly as accurate as DDG, but takes longer to calculate.
Warning: DDG runs local Relax with your point mutations for computational ease. This allows us to run a large number of point mutations in the same time a full Relax would take. However, this is known to make the method less predictive when performing small to large point mutations. For example, an Alanine to Tryptophan will almost always score as unfavorable. However, this mutation can occasionally be favorable, but it would have required global Relax to find that optimal conformation that accommodates the Tryptophan.
EXAMPLE OF HOW DDG CAN BE USED: Predicting stabilizing mutations for the p53 core domain
For this example a crystal structure of the p53 core domain, 2OCJ, was loaded into CAD where Prepare was run once followed by 100 repeats of Relax. Then the lowest energy structure was used to run DDG, with all possible mutations allowed for the entire structure.
The results in this example showed that only 23 of the 194 residues had point mutations with at least one DDG < -1.0 kcal/mol (shown in blue below).
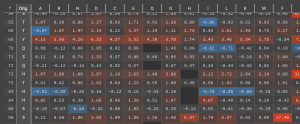
These 23 positions can be used to guide Design runs in order to stabilize the structure. Of the 23 positions, there were 65 point mutations that have DDG < -1.0 kcal/mol.
Always avoid making mutations at locations that would affect function. In this case, five of the positions were in the DNA binding region which left only 18 positions with 58 mutations. If the original residues had been included in Design, that would have made over 18 billion possible sequence permutations.
It would take years of calculation to properly sample them. However, if you run Design for three to six positions at a time, you can split it into four Design jobs. Then the best sequence sets from these jobs can be used to run a full protein Design. This will take just a few hours per Design and the final answer will be almost as good as the larger calculation, because local interactions have the strongest influences on mutations.
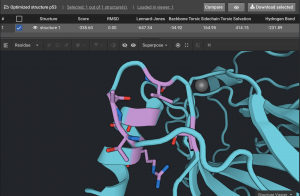
In this example with p53, there were four clusters of residues as shown below. Each cluster consists of residues that are within a single secondary structure or that are in a chain of interacting residues. The cluster shown below has six residues (in purple) and has 14 mutations. This is a very tractable Design case.
For each one of the four sets Design can be run up to 1,000 repeats. One of the four Design jobs only had 16 possible permutations and 100 repeats were run. Running 100 is usually sufficient to see the potential favorable sequence space, but upping it to 500 or 1,000 can find some more diversity. The Sequence Logo for the a Design run is shown below.
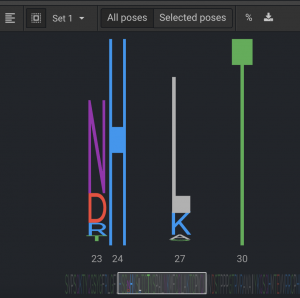
From the Sequence Logo, you can see that positions 23 & 27 have several potential mutations and 24 & 30 only have the original residues present. It is better to trust Flex or Relax Design more than DDG because it allows more conformational sampling, but vanilla Design has less because it only moves side chains.
After each Design set is run, look at the Sequence Logo to see what residues were most often selected. From the Sequence Logo above, select the seven possible mutations at positions 23 & 27 and keep WT for 24 & 30. The same protocol is done for the other three design sets for this protein so that all desired positions can have design done simultaneously, using this smaller, more informed mutation set. In this case, there were 48 permutations. Then Relax Design was run with 500 repeats. The sequences of 80% of these converged on a single sequence. The new sequence had lower energy by 7.9 REUs, which is a significant improvement and had mutations at seven positions.
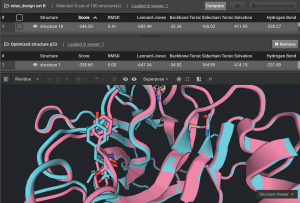
Above is the original structure in blue and the stabilized mutant in pink in cartoon mode. The mutation positions are shown in sticks.
Additional stabilization may be possible by starting with the stabilized structure and repeating the process. Starting with a new DDG calculation. Then using the results to guide another round of design. You can see that the backbone is nearly identical, but the slight backbone shifts and new sequence may find a few more mutations to add to the design process.