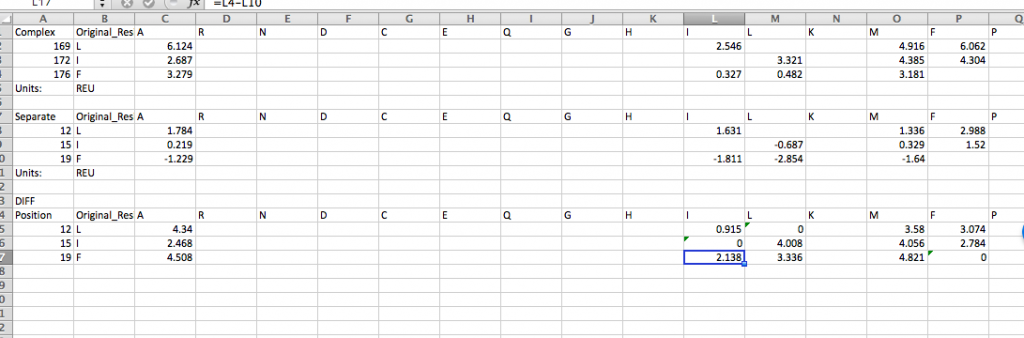
INTERFACE DDG WORKFLOW
The free energy of an interface single point mutation (change in binding free energy) can be calculated using this workflow. Here we will look at mutations on the alpha-helical peptide BHRF1 in complex with the Bim BH3 domain, 2WH6.
STEP BY STEP:
1.) Load 2WH6 in Bench.
2.) Run Prepare on the structure, in our example here that is Prepare 12.
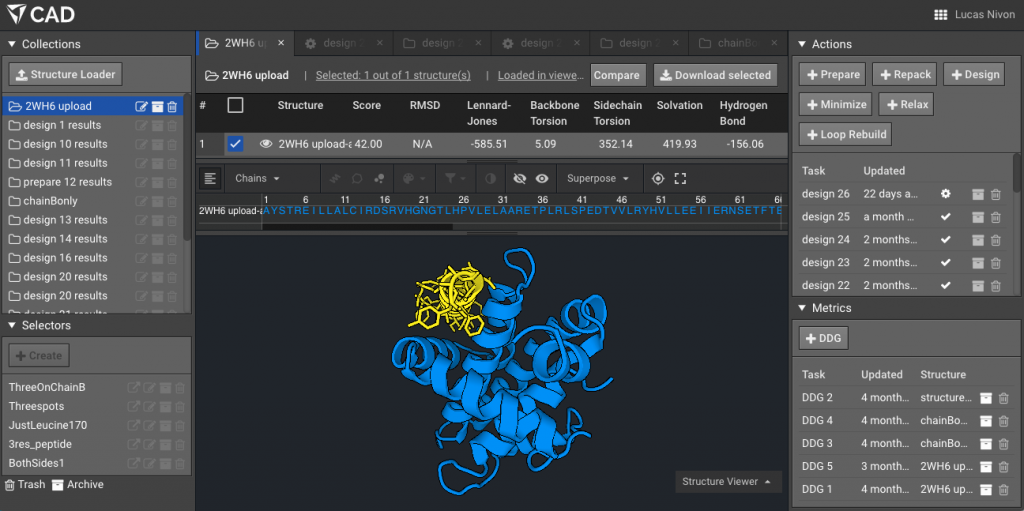
3.) Optional but helpful: show the two chains using “Chain” selector mode, here the peptide is in yellow.
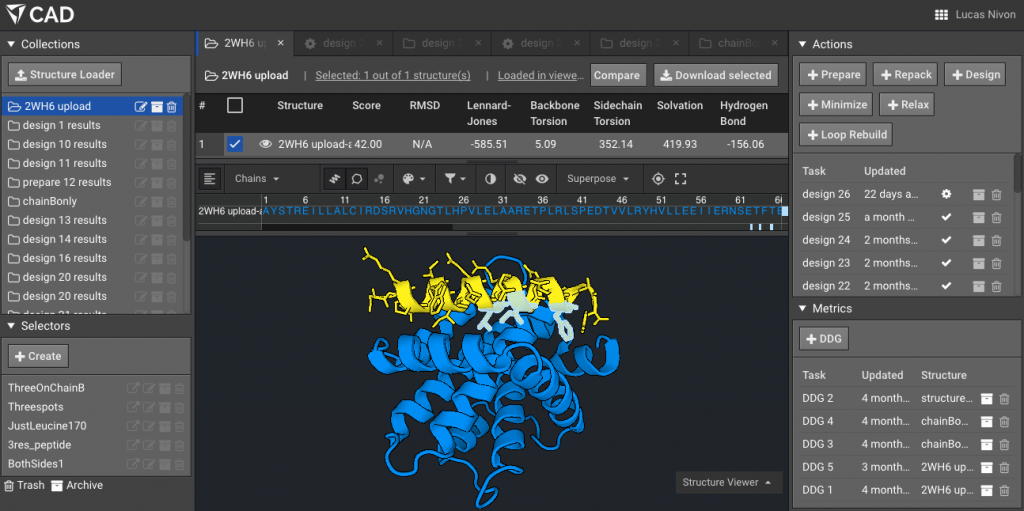
4.) Make a Selector with the residues of interest (the ones that will mutate), here for example the three residues on the peptide side of the interface that we are interested in are L169, I172, F176 (note that Bench uses Rosetta numbering, where all residues are numbered sequentially from 1 regardless of chain). Here we have named that Selector “Threespots”.
5.) Run DDG on those residues of interest — these scores are your “complex” scores. If you need more detail, follow this tutorial on how to run DDG. Here we have put in mutations to AILMF at each of our positions of interest, and the Metric is called DDG8.
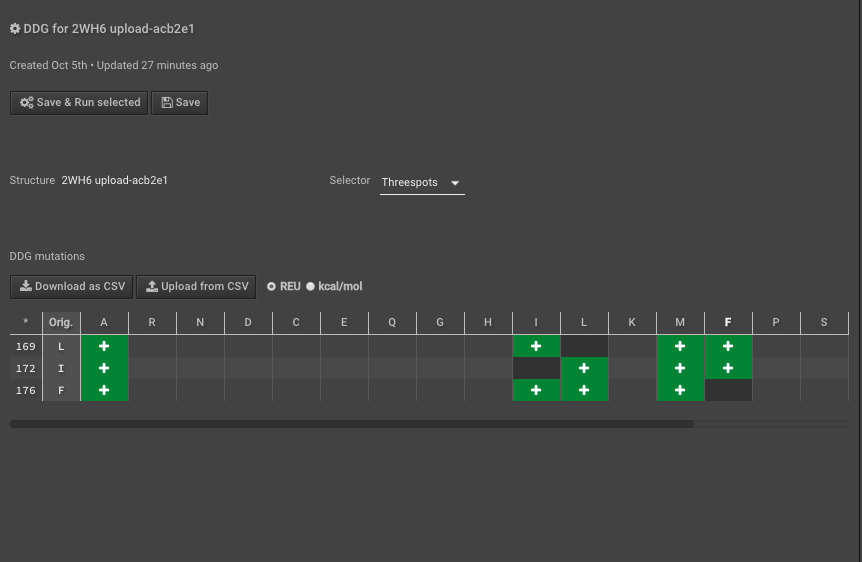
6.) When this is done record the results either separately or just “Download as CSV”, here I will use the CSV to do multiple calculations easily.
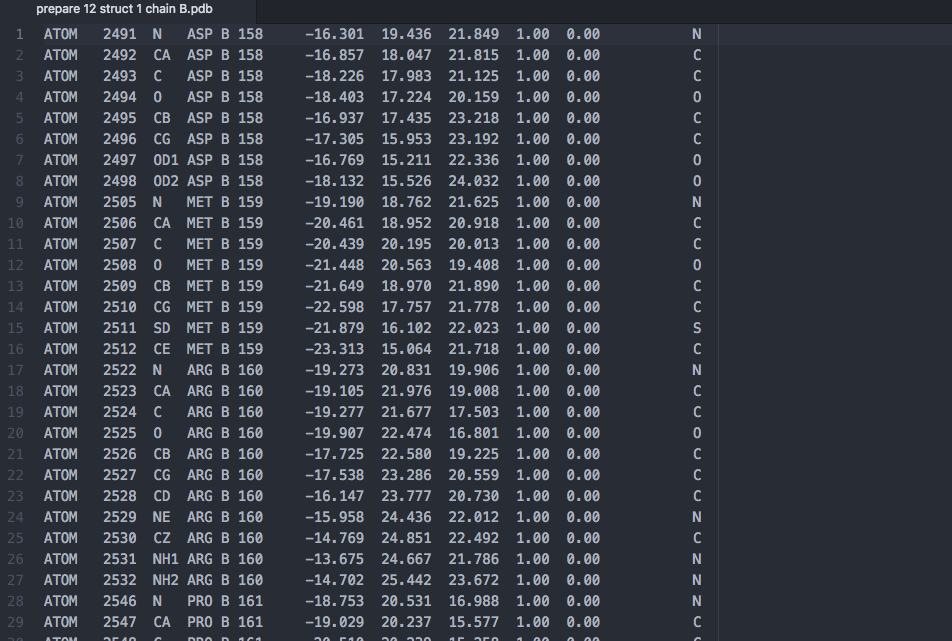
7.) Download the original prepared structure, here that is prepare 12. Copy and save that file under a new name. In a text editor remove the non-mutating (constant) chain. In this example chain A is the target that is not mutating and chain B has our mutations, so I will simply delete all of the lines for chain A in a new text file saved as “prepare 12 struct 1 chain B.pdb”. Here we are showing the top of that new text file after deletion.
8.) Load that mutating-chain-only file into Bench from your file system. Here that is loaded as “chainBonly”. Now we are effectively going to repeat steps 3-6 for this chainB only molecule.
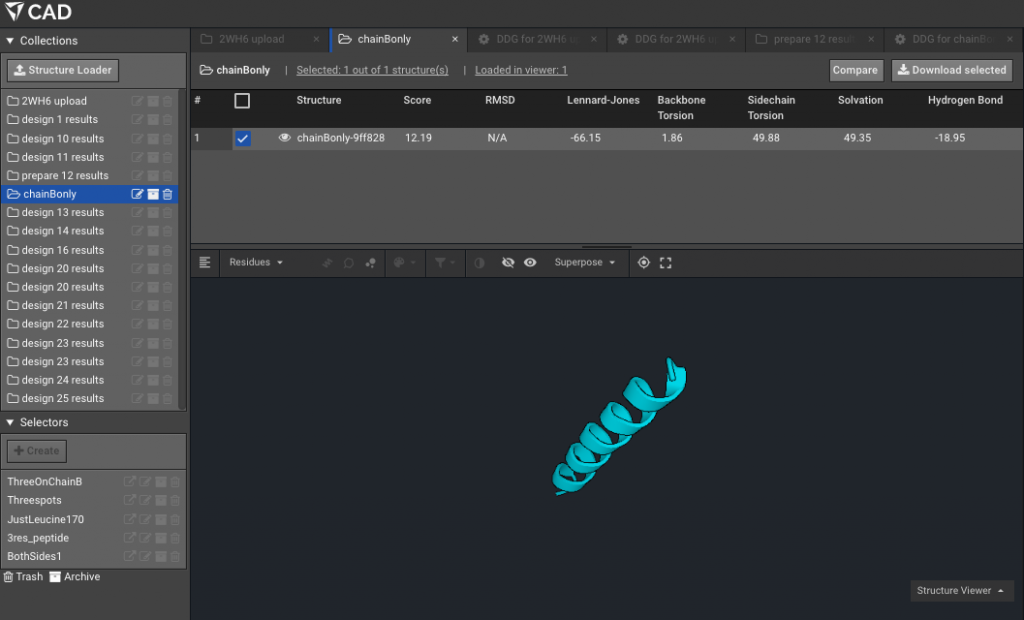
9.)Make a Selector with the residues of interest, the ones that are mutating. Here for example on chainB, with the new numbering due to Rosetta numbering, our residues of interest formerly 169, 172 and 176 are now 12,15 and 19. Here we have named that Selector “ThreeOnChainB”.
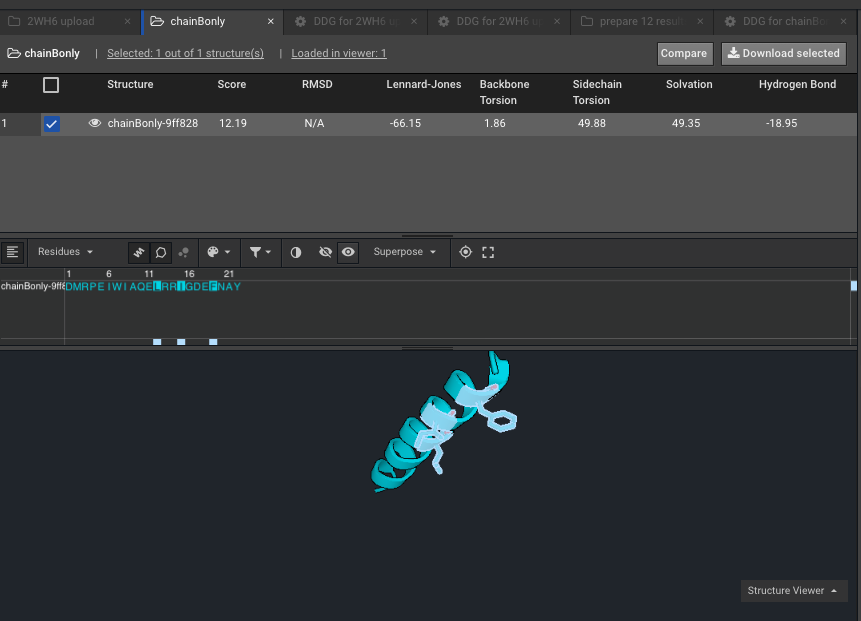
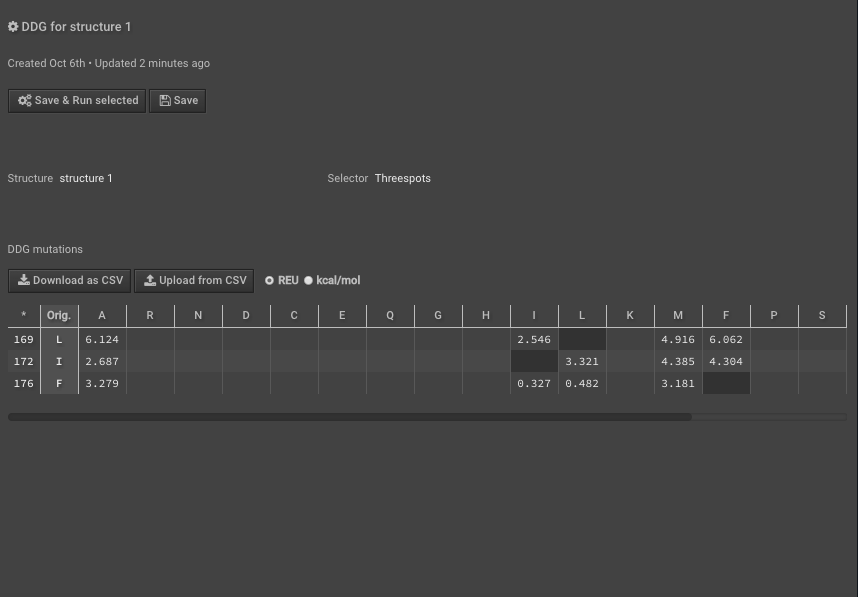
10.) Run DDG on those residues of interest noting that the residue numbers are different now because of Rosetta numbering — these scores are your “separate” scores. Here we have put in mutations to AILMF at each of our positions of interest and the Metric is called DDG7.
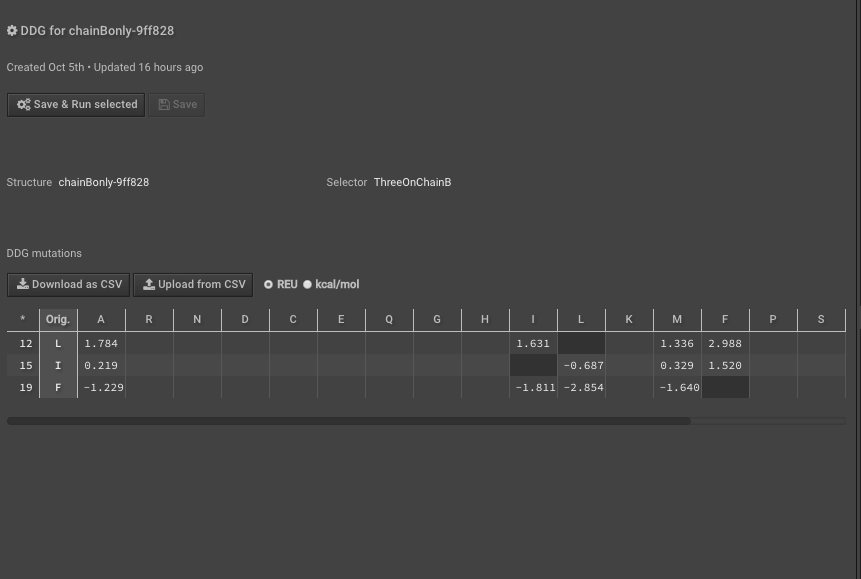
11.) When this is done record the results either separately or “Download as CSV”, here we are downloading the CSV.
12.) Last, to calculate the interface_DDG for any mutations, calculate the difference of the individual values, so interface_DDG = complex_score – separate_score. That is simple to do for many mutations at once using the CSV files you’ve downloaded above. For example, here we calculate the interface_DDG(L12I) = 0.915, and all of the mutations have a positive interface_DDG in this example.