INTRODUCTION
Many design tools have been developed by Rosetta labs and the three most refined methods have been incorporated into Cyrus CAD for easy use. These tools (Design, Flex Design, and Relax Design ) have been well-tested in the Rosetta labs where they were created and have been successfully used to generate structures with improved stability, solubility, and affinity. Additionally, structures have been designed that alter catalytic activity, specificity, and have even created folds not found in nature. The three design tools in CAD are useful for different design goals and are defined in the next section.
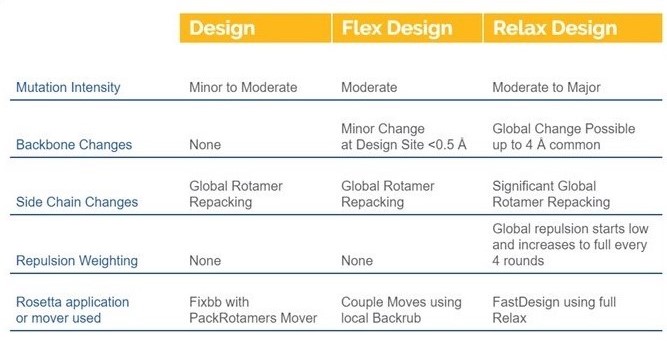
Design, Flex Design, and Relax Design take a protein structure and allow you to select locations in the structure where you would like it to sample possible mutations. You can allow all 20 amino acids or select fewer possible residues to be changed during the molecular simulation. Each position can have a different set of possible mutations. While all three methods allow you to make as many mutations as you like, each of them will run the simulation very differently and is likely to come up with very different solutions to the same case. This chart highlights some of the major differences. For instructions on how to run a design job click here:
DESIGN
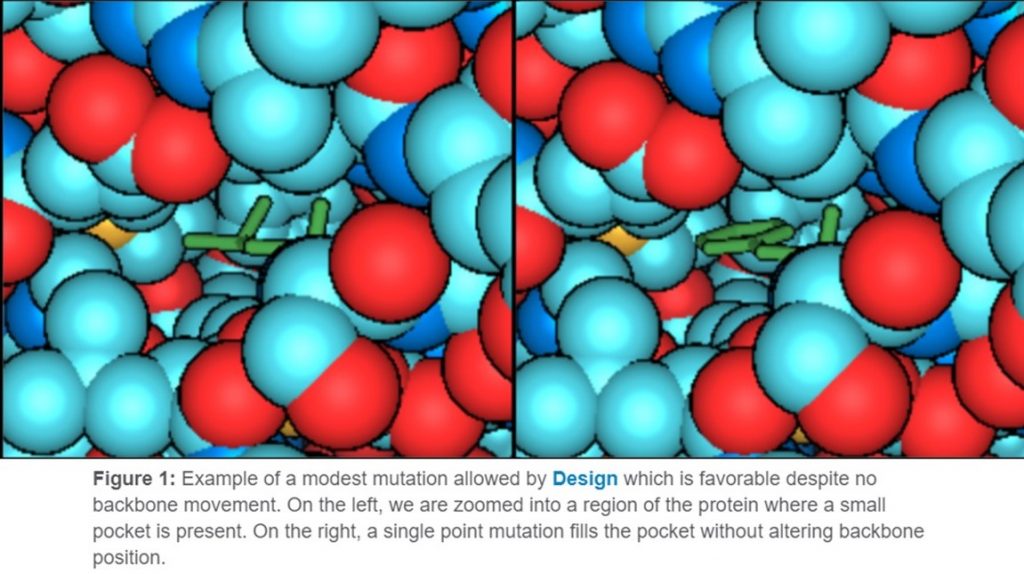
Design is the most conservative of the three methods. All three methods are able to create mutations that affect stability, affinity, specificity, and solubility. Though the level of change you can expect from each method varies. Design will result in more minor changes because there are no backbone changes allowed during the simulation. So, when a mutation is sampled, it is highly constrained. For example, a buried hydrophobic amino acid that mutates to a polar residue will be highly unfavorable. But a minor change to another hydrophobic that better sterically fits would be more favorable (Figure 1).
However, the conservative nature of the mutations can be advantageous is many situations. For example, if you are working with a protein and you want to maintain the fold in order to maintain active site or interaction site conformation, you are more likely to preserve these features by using Design. Often users need a sequence change in order to alter immunogenicity, post-translational modification sites, or propensity for aggregation, but do not want other changes in the structure. In this case, the fold could be maintained while looking for other sequences that are still energetically favorable in that fold but have a more favorable sequence.
The Design protocol samples side chain mutations at the design sites and samples side chain orientations of all positions in order to find the most energetically favorable orientations given a fixed backbone. The Cyrus Repack action does that same sampling of side chains. So Design is essentially running Repack while allowing mutation at the design sites.
- ORIGINAL PAPER FOR THE DESIGN METHOD:
Kuhlman B, Baker D. Native protein sequences are close to optimal for their structures. Proc Natl Acad Sci U S A. 2000 Sep 12;97(19):10383-8. Erratum in:Proc Natl Acad Sci U S A. 2000 Nov 21;97(24):13460.
- EXAMPLES OF THE DESIGN METHOD:
Kuhlman B, Dantas G, Ireton GC, Varani G, Stoddard BL, Baker D. Design of a novel globular protein fold with atomic-level accuracy. Science. 2003 Nov 21;302(5649):1364-8.
Mills JH, Sheffler W, Ener ME, Almhjell PJ, Oberdorfer G, Pereira JH, Parmeggiani F, Sankaran B, Zwart PH, Baker D. Computational design of a homotrimeric metalloprotein with a trisbipyridyl core. Proc Natl Acad Sci U S A. 2016 Dec 27;113(52):15012-15017.
FLEX DESIGN
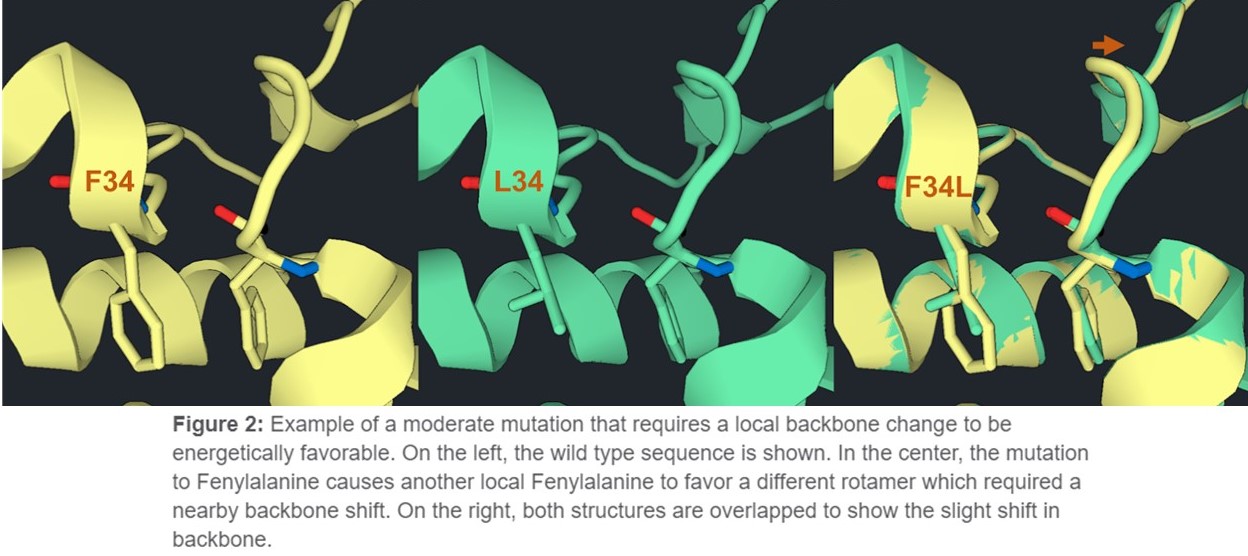
Flex Design is the more intermediate design tool in terms of aggressiveness. It is less constrained than Design because it allows some backbone changes to occur, but it only samples these changes at locations that you define as mutatable. So, your global fold will remain the same. This allows a bit more conformational sampling which allows slightly more aggressive mutations to occur. For example, we showed a hydrophobic mutation that filled a gap with Design. But if Flex Design made a mutation to a larger side chain in the protein core, a slight shift in the backbone might accommodate the change (Figure 2).
The protocol is a Monte Carlo (MC) optimization which iteratively samples new conformations to minimize in terms of Rosetta energy (more on Rosetta energy). At each iteration, there is a random backbone position that is allowed to be perturbed. The perturbation is called a Backrub. This involves rotation along the axis of the backbone at three consecutive amino acids so that the center residue is the potential mutation site. Then the mutatable site is randomly mutated to one of the allowable mutations (that can include the wild type). Then all orientations are sampled for that side chain to find the most energetically favorable one. This is followed by global side chain optimization. This is done iteratively to find the most energetically favorable mutations and conformation.
- For more information on Backrub:
Davis IW, Arendall WB 3rd, Richardson DC, Richardson JS. The backrub motion: how protein backbone shrugs when a sidechain dances. Structure. 2006 Feb;14(2):265-74.
- For more information on Flex Design (called Coupled Moves), see its original publication:
Ollikainen N, de Jong RM, Kortemme T. Coupling Protein Side-Chain and Backbone
Flexibility Improves the Re-design of Protein-Ligand Specificity. PLoS Comput
Biol. 2015 Sep 23;11(9).
- For more Flex Design (Coupled Moves) papers that show its utility and predictive power:
Humphris EL, Kortemme T. Prediction of protein-protein interface sequence diversity using flexible backbone computational protein design. Structure. 2008 Dec 10;16(12):1777-88.
Smith CA, Kortemme T. Backrub-like backbone simulation recapitulates natural protein conformational variability and improves mutant side-chain prediction. J Mol Biol. 2008 Jul 18;380(4):742-56.
Smith CA, Kortemme T. Structure-based prediction of the peptide sequence space recognized by natural and synthetic PDZ domains. J Mol Biol. 2010 Sep 17;402(2):460-74.
Smith CA, Kortemme T. Predicting the tolerated sequences for proteins and protein interfaces using Rosetta Backrub flexible backbone design. PLoS One. 2011;6(7).
RELAX DESIGN
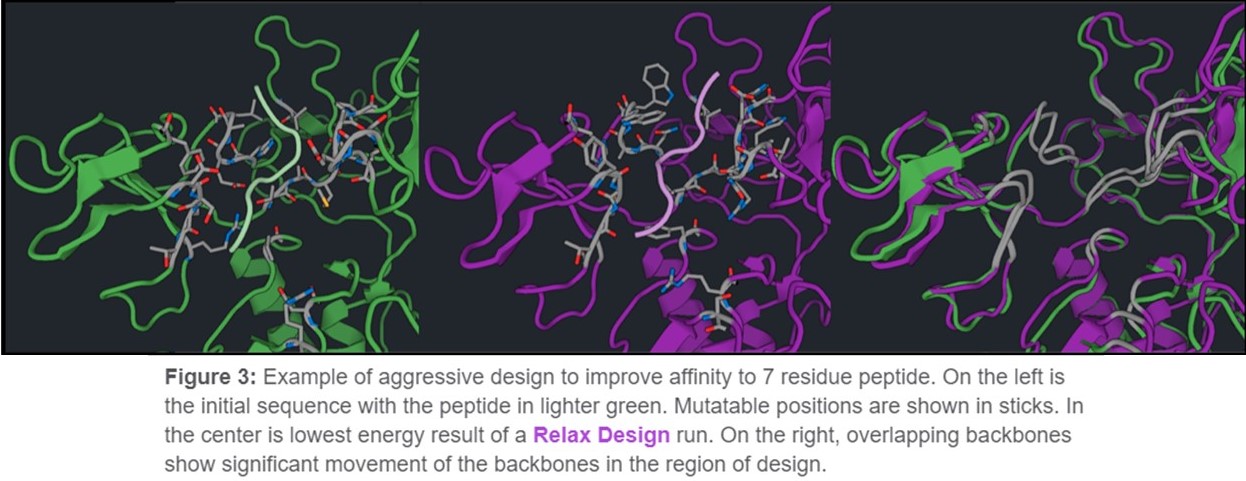
Relax Design is the most aggressive design tool in CAD. It allows backbone changes for the entire structure. This is ideal if you are looking to make mutations that would require movement beyond the mutation site to energetically accommodate the change. Many design mutations could require conformational changes outside of the design region in order to accommodate more aggressive mutations. It is more likely to be necessary when making mutations in a large area, and in practice is commonly used for de novo design of entire proteins or of protein/protein binding interfaces at all positions. For example, in Figure 3 we allowed residues interacting with a seven residue peptide.
The backbone changes are made with the protocol called Relax (as the name implies). This tool makes a perturbation at a random backbone location in the protein. This is followed by optimization of side chains to potentially accommodate the perturbation.
- Examples of the Relax Design method:
Nivón LG, Moretti R, Baker D. A Pareto-optimal refinement method for protein design scaffolds. PLoS One. 2013;8(4).
- For more information about Relax:
Conway P, Tyka MD, DiMaio F, Konerding DE, Baker D. Relaxation of backbone bond geometry improves protein energy landscape modeling. Protein Sci. 2014 Jan;23(1):47-55.
RUNNING DESIGN WITH A PSSM
A protein Position Specific Scoring Matrix (PSSM) takes the alignment of your protein to a list of homologs and counts the amino acids found at each position for the homologs. This is an easy way of measuring evolutionarily related mutations accepted at each position. This can be used to guide protein design because mutations found in homologs are likely to maintain structural integrity and function. You can create a PSSM for your protein, then use it as a list of potential mutations for design in Cyrus Bench. For instructions on how to create a PSSM click here.