BACKGROUND
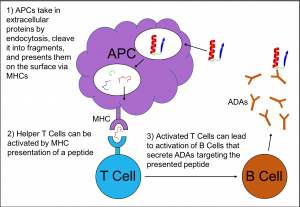
Immunogenicity is the propensity of a molecule to provoke a response by the immune system. In many cases this is the desired behavior of the immune system, recognizing foreign objects and targeting them for neutralization or removal. However immunogenicity is a major problem for design of biologic therapeutics because a molecule intended to treat disease may be recognized as foreign by the immune system and be targeted for degradation and removal. Immunogenicity can be prevented by redesigning the drug in order to avoid the very first step in the cascade of signals. Epitope Scan is a tool to predict whether a protein will be targeted by the immune system. It focuses on a peptide-binding receptor called Major Histocompatibility Complex class II (MHC II). MHC II is responsible for recognizing “foreign” peptides and leads to activation of the immune system to create antibodies against those peptides, or in the case where the peptide is part of a biologic drug, anti-drug antibodies (ADA).
MHC IIs are found on Antigen Presenting Cells (APCs). APCs absorb extracellular proteins, cleave them into peptide fragments, and load these peptides into MHC II. They translocate these peptide-receptor complexes on the APC cell surface and “present” the peptides to T Cells. If a T Cell receptor recognizes the peptide in the MHC II receptor as a “non-self” peptide, the T Cell becomes activated. Activated T Cells go on to trigger the activation of memory B Cells. These B cells secrete ADAs that bind, disrupt, and eliminate the original foreign protein, our biologic drug of interest. These ADAs may simply block and eliminate the target drug, or in more severe cases can lead to systemic allergic reactions, anaphylaxis, or even initiate autoimmune disorders.
OVERVIEW
Our Epitope Scan tool will take your protein and identify linear regions of the sequence with the potential to activate the immune system. This is done by predicting which peptide fragments in the input protein are most likely to bind MHC II receptors with high affinity. It is based on the work of Indigo King (1), who also implemented it in Cyrus CAD. The goal is to identify regions that could activate the immune system by binding to MHC II receptors, then create mutations in these areas to prevent MHC II receptor binding, evading immune cell recognition while preserving the protein’s structure and function.
This can be done manually by running the Cyrus prediction algorithm and then redesigning the likely immune epitopes using CAD, or using a fully automated approach. As of May 2018 the current version can be used to carry out the two step approach prediction and design. In a future release in 2018 we will also offer a single turnkey protocol in order to facilitate simultaneous prediction and elimination of potential T Cell epitopes while maintaining proper folding and function of the target protein.
Peptides that are recognized by MHC II have been extensively characterized and have been curated into a database of epitopes the IEDB. King used the database’s experimentally-derived peptide affinity of MHC II alleles, combined with machine learning to create a scoring mechanism that is predictive of immune system activation. It scans 9 residue regions to predict the affinity of each MHC II allele.

RUNNING EPITOPE SCANS
Click on the collection folder containing your protein on the left in order to bring it into the center window. Check the box to the left of the structure ID ![]() . Then just click
. Then just click ![]() and it will run a full scan. As the data is being processed, it will say Running.
and it will run a full scan. As the data is being processed, it will say Running.
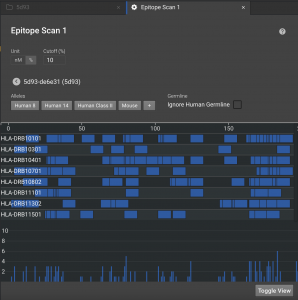
Once finished, click on the structure name to see a visual display in the default view. There are 8 HLAs shown, but you can expand what is shown by clicking ![]() to load 14 alleles or
to load 14 alleles or ![]() to load all 26 in our database. The blue bars in each row indicate the 9 residues windows that have affinity below the cutoff. The chart at the bottom show the number of alleles that have affinity for that window.
to load all 26 in our database. The blue bars in each row indicate the 9 residues windows that have affinity below the cutoff. The chart at the bottom show the number of alleles that have affinity for that window.
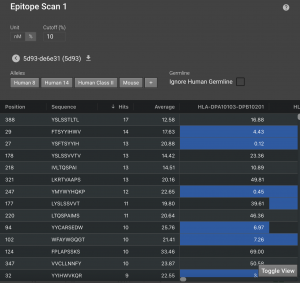
Click Toggle View to bring up a table that will show all the 9-residue regions that were analyzed. It lists their position, sequence, the MHC allele and its predicted IC50 for the sequence. Anything under your cutoff will be highlighted in blue. By default, the IC50 cutoff for weak binders is set to 500 nM. The cutoff for strong binders is often considered to be 50 nM. You can also set the cutoff by % rank. For each MHC allele prediction model, we predicted MHC binding affinity to each of 100,000 random peptides from open reading frames in the human genome. Any time a new peptide sequence is scored, its rank within that reference genome set is calculated (e.g. a rank of 1% means that the target peptide is in the top 1% of possible epitope scores for that MHC type).
By clicking Hits, you can sort by the number of hits, meaning the number of alleles that have affinity for each 9 residue window. You can return to sequential sorting by clicking Position.
By default, it will show you all 26 MHCs used for analysis, but it is common practice to just look at the top 14 or top 8 MHCs. These are usually representative of immunogenicity, but not comprehensive.
RANK PERCENTAGE:
The most informative way to analyze the results is to look at Rank percentage. This is a normalized measure of epitope probability (e.g. a rank of 10% indicates that this epitope is in the top 10% *highest* predicted epitopes for this allele type). Rank percentage is more useful than IC50 for comparing the immunogenic potential of a given epitope sequence between different HLA allele types because certain alleles tend to bind more or less strongly to epitope peptides in general even though they may have equal immunogenic potential. Rank percentage is calculated by comparing the IC50 binding prediction to a reference set of prediction scores from 100,000 random 15mers from all open reading frames in the human genome. The rank value reported is calculated as 100% * (1.0 – rank_fraction_in_sorted_ref_scores ).
Toggle the results from nM to % at the top of the Epitope Scan screen:
By default, it will show only the epitopes predicted to bind in the top 10% in comparison to the reference set, though this value can be shifted. You can further sort the remaining epitopes by the # of alleles that have predicted affinity under your cutoff, average affinity, or by individual alleles. Simply click the desired column header to sort.
REDESIGN PROTEIN to A SEQUENCE BASED ON EPITOPE SCAN
In order to run design, we recommend choosing one region at a time that is within a single secondary structure. Alternatively, you can choose a set of residues involved in an interaction between two secondary structures. Start with a diagnostic design run. Then use the information learned from this run to do the actual design. In the first run, allow mutations that are relatively conservative, meaning they have similar hydrophobicity and size. Avoid Cysteines and Prolines that aren’t already in the structure because they tend to cause significant structural changes. Try not to mutate positions that are already Cysteines or Prolines. Run design with 1,000 repeats in order to get enough sampling of potential mutation sets.
After you have run the diagnostic design, look at the Sequence Logo to see which residues are highly prevalent in the design positions. Use this to guide another design run with 1,000 repeats. Select the mutations that are common in the diagnostic run as allowable mutations for the next design run. Once this is done, run epitope scan on each of the different resulting sequences (there should be a lot of structures with the same sequences). Select the sequences that have improved Epitope Scan values and use these as starting structures for more design in another region of the protein that has an Epitope Scan value below your cutoff. You should also be analyzing designed structures for structural changes at each step. You can use Compare and Superpose to see if there are significant differences.
Warning: Conservative amino acid mutations are more likely to have similar immunogenicity. So regions where you know that structural changes are permissive, you are more likely to evade the immune system with disparate mutations.
In order to see which residue in a 9 amino acid window is likely to activate the immune system, go to http://www.cbs.dtu.dk/biotools/MHCMotifViewer/Human_alleles.html.You can look at each MHC II allele to see the Sequence Logo representation for the epitope that it binds. So, if you want to mutate a region that is under the cutoff for allele DRB10101…
Click the ![]() at the above website. Then click
at the above website. Then click  . This can be used to guide mutations selection as described below.
. This can be used to guide mutations selection as described below.

For more information on Design: Click here
For more information on Epitope Scan:
- King C, Garza EN, Mazor R, Linehan JL, Pastan I, Pepper M, Baker D. Removing
T-cell epitopes with computational protein design. Proc Natl Acad Sci U S A. 2014
Jun 10;111(23):8577-82.