Minimize is a quick way to find the optimal conformation of your protein in the near vicinity of the structure’s current orientation. This will find the local energetic minima, but not the global minima. It is often used as a last step during structure optimization.
RUNNING MINIMIZE
Select the folder that contains the structure or structures that you want to Minimize.
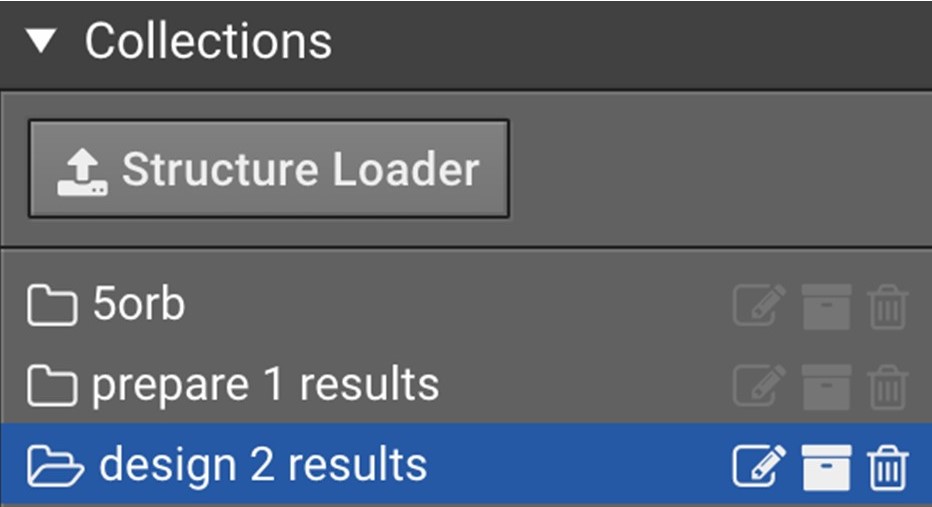
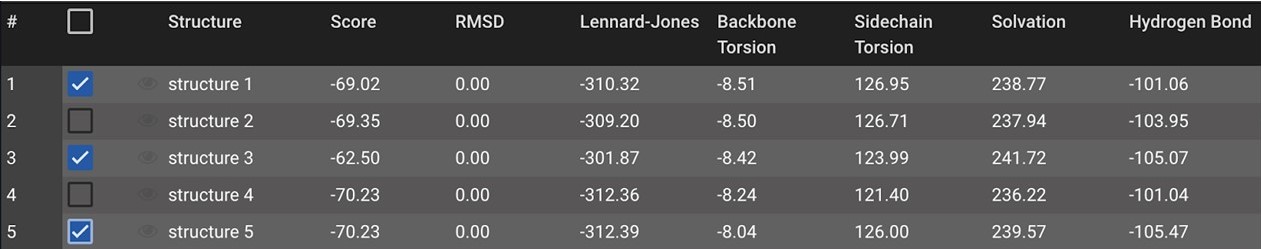
This will bring the structures in that folder into the center window. Select all the structures that you want to Minimize by clicking in the box to the left of their names.
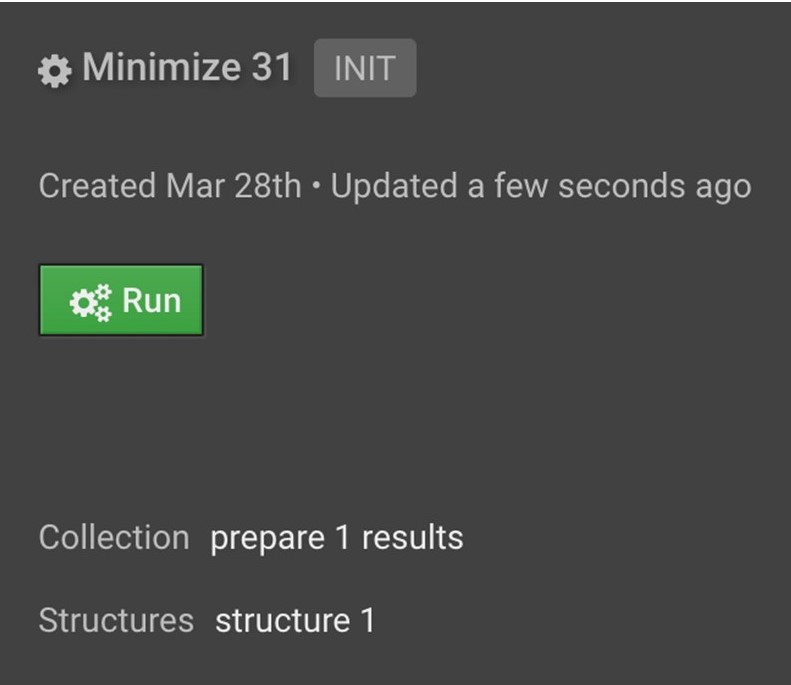
Next, click the ![]() button on the top right under Actions to initialize Minimize in the center window. Click Run.
button on the top right under Actions to initialize Minimize in the center window. Click Run.
WHAT DOES MINIMIZE DO?
This is Rosetta’s default Minimizer which, unlike most of our other optimizations, is not a Monte Carlo method. It is a gradient-descent minimization of Rosetta energy. This is a deterministic method so it isn’t necessary to run multiple minimizations like is recommended for Relax because repeats will just find the same final conformation. The terms that make up the Rosetta energy function are written in a way that allows us to calculate their first derivative in terms of spatial degrees of freedom. You may remember from calculus that the first derivative is the slope of a curve, which in this case will tell you how to change the conformation of your protein in order to decrease the energy. Though, we are making tiny conformational steps downhill iteratively until it can no longer find a slope downward.
In mathematical terms, it is a Quasi-Newton, line search with the Armijo–Goldstein condition using partial derivatives of the Rosetta energy function. This is a variant of the Broyden-Fletcher-Goldfarb-Shanno (BFGS) gradient-based minimization.
The Rosetta terms for this are default for minimize (dfpmin), and are:
-run:min_type lbfgs_armijo_nonmonotone
-run:min\_tolerence 0.001
For more information about protein structure in terms of energy, see this video.