OVERVIEW
Loading a structure file into Bench is a two step process.
Step One: Select which components of the structure file you wish to be included.
Step Two: Bench processes the file contents to define all molecules in a way that allows Rosetta to understand the physics of everything present.
Importing a structure into Bench can be a very simple or complex process depending on what is present in your file. When only loading a protein, there is not much to consider, but the handling of non-protein molecules requires some potentially complex, but important steps.
The conformation and sequence preferences of a protein can change significantly when bound to another molecules. So, the ability to model proteins with their non-protein binding partners is hugely important. Rosetta is excellent at doing so, but it takes a great deal of work to create the files that will provide the physics-based descriptors for each ligand in order for Rosetta to model them properly. This is a cumbersome task in Rosetta. Unique to Cyrus Bench is the ability to automatically and reliably parameterize non-peptidic residues. The automated parameterization process integrates characterization tools from Rosetta with additional tools from the OpenEye small molecule toolkit. Open Eye is a leader in the field of small molecule cheminformatics and their toolkits are widely used by small molecule bioinformaticists.
LOADING A PROTEIN WITH SMALL MOLECULES
When you load a pdb file into CAD that contains non-protein elements, those elements can be processed and included in the system. Rosetta does not have an automated way to understand non-proteins, so they require expert handling. Rosetta already has extensive software for automated characterization of a protein. It can quickly load a pdb and create a profile that defines it’s chemistry. As complex as proteins are, they are all composed of the same 20 amino acids which have well characterized physics. The chemical space required to define non-peptidic molecules is much more expansive. Defining the physics of non-peptidic molecules requires analysis of the topology and subsequent determination of atom types, partial charges, and rotomeric preferences.
Important Note: We currently do not allow you to keep DNA, or RNA. We also limit the protein to the 20 canonical amino acids. We convert non-canonical amino acid residues to their closest canonical parent. We will eventually allow DNA, RNA, and non-canonical residues. These can be very important, but further code development is necessary to include proper modeling. Because Rosetta/Bench uses an implicit solvent water model, most/all crystallographic waters should typically be discarded. On occasion, a water serves a specific structural purpose, such as bridging interactions between a small molecule ligand and the protein. In that case, you may wish to include a very limited number of explicit waters.
HOW TO LOAD A STRUCTURE WITH SMALL MOLECULES PRESENT
You can load a structure that has a small molecule with the Structure Loader:
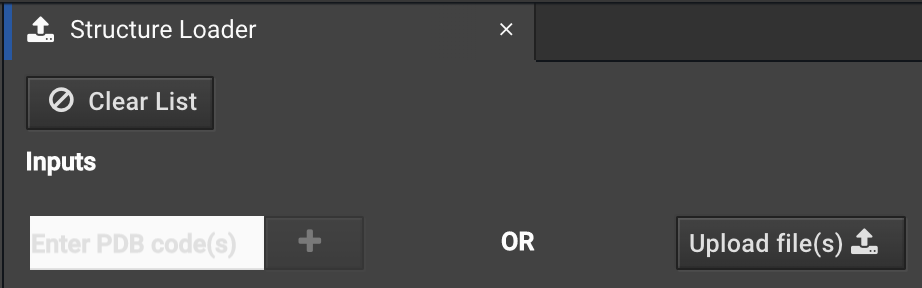
You can enter your PDB ID or load a file from your computer as usual.
In the next step, you will be asked which non-protein molecules from the file you wish to keep. By default, all protein, DNA, and RNA residues are kept and other atoms are shown as things you do NOT want included. However, you can add back or remove anything.
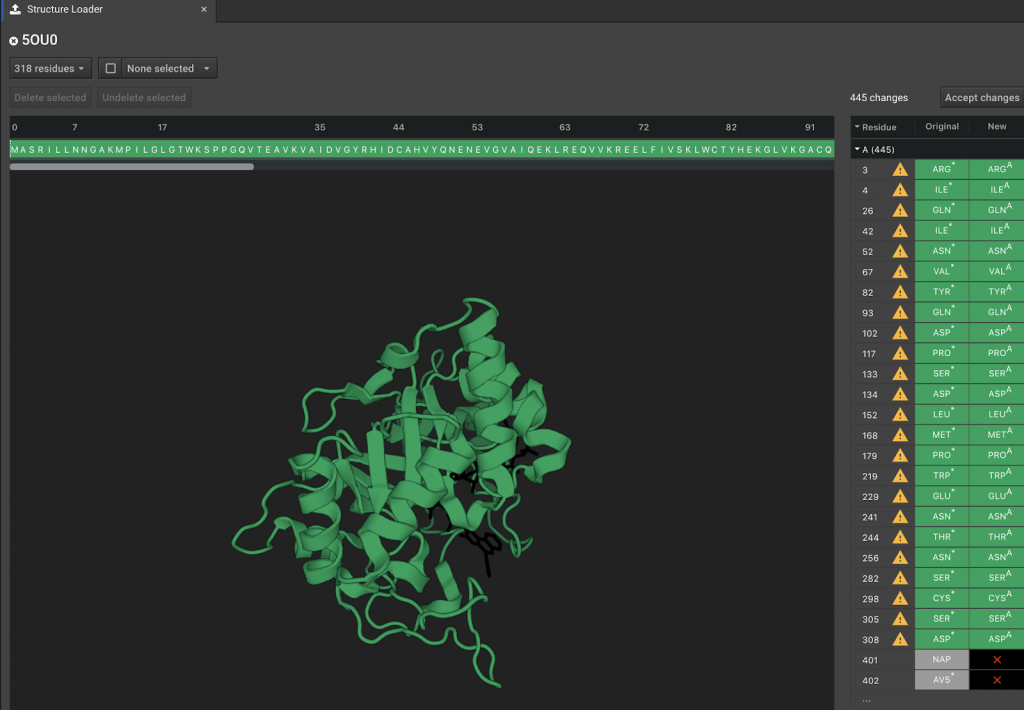
In the top of the tab window, there are 3 drop down buttons. In this example, the first reads ![]() . (The number will change depending on how many residues are present.) Clicking this button reveals a summary of what residues and molecules are visible in the Structure Viewer. Any changes you make in this box will change the appearance in the workspace, but don’t by themselves delete or undelete elements of the system. Two examples are shown below.
. (The number will change depending on how many residues are present.) Clicking this button reveals a summary of what residues and molecules are visible in the Structure Viewer. Any changes you make in this box will change the appearance in the workspace, but don’t by themselves delete or undelete elements of the system. Two examples are shown below.
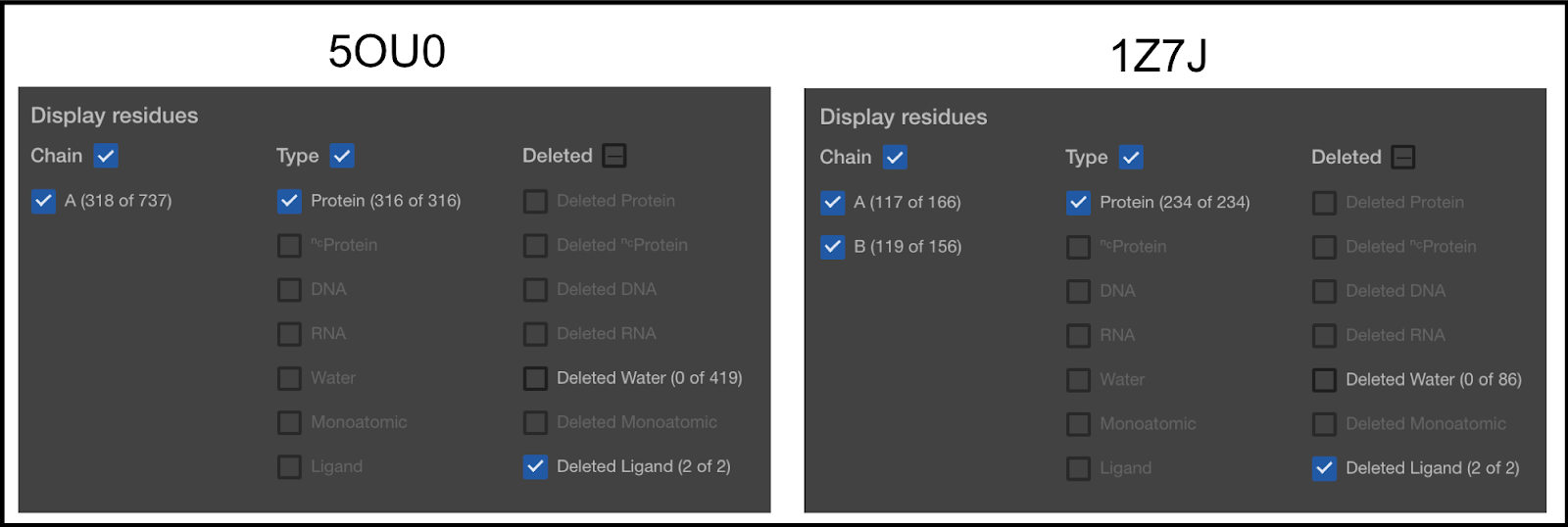
Anything that has its box checkmarked will be visible, even if it is set to be deleted. Anything not checkmarked will not be visible, even if it is set to be included. In the first column, you can see that 5OU0 has 1 chain (A) and 1Z7J has 2 chains (A and B). Later in this article, you will be shown how to remove chains.
In the second column, the types of molecules selected are shown. You can see that the type of molecules currently selected is only the Protein molecules. Our default mode is to only keep the protein, DNA, and RNA. The second item in the list (NC Protein) refers to non-canonical proteins. We currently convert all NC Proteins to canonical. So, for example, if you have Selenocysteine on your protein, it will be converted to a Cysteine. The others are self explanatory, except for Ligand. This term is used for anything else.
The third column lists the things currently removed from the Structure Viewer.
As you add things to be included in the final structure load, things will become visible in the Viewer. That will be indicated in this window as well.
The next button, ![]() will allow you to alter which things you have currently selected.
will allow you to alter which things you have currently selected.
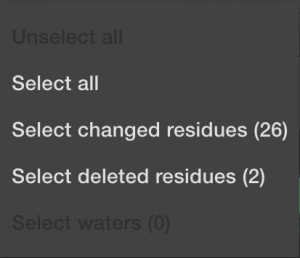
This can speed things up if you know you want to include everything. Or deselect everything if you know you only want to choose a few things manually. Or if you want to select all the things that have been automatically changed during import, meaning the non-canonical amino acids. And finally, you can select all the deleted items, typically the mono-atomic or ligand molecules. Again, DNA and RNA will not be allowed until the next software upgrade.
The third button on the top says ![]() by default, but can be switched to
by default, but can be switched to ![]() . This is helpful if a crystal includes multiple copies of the relevant protein or protein complex in the unit cell. When in
. This is helpful if a crystal includes multiple copies of the relevant protein or protein complex in the unit cell. When in ![]() mode, you can add or remove whole chains in the Structure Viewer. Select the elements you want to modify, then click
mode, you can add or remove whole chains in the Structure Viewer. Select the elements you want to modify, then click ![]() or
or ![]() .
.
The workspace, which appears below, presents a visual view of the molecules in the PDB file. Clicking on various elements of the structure, along with using the buttons above, provides the user with another way to add/delete included elements. Deleted elements of the system appear black.
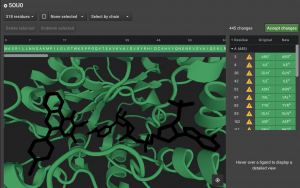
Above the Structure Viewer is the sequence. Mono-atomic, water, and Ligand items appear after the protein. Any deleted elements are given the single letter x in the sequence. If you undelete any of these elements, it becomes z in the sequence or w for water. Any protein residues that are deleted will also be represented with an x.

Finally, the panel to the right of the Structure Viewer lists the individual items that are currently deleted or that have been altered.
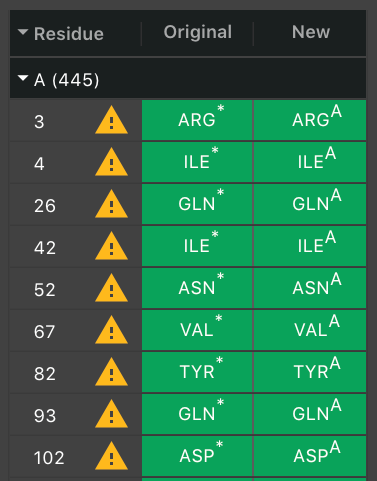
They appear in order of their residue number. The next column will have ![]() if a residue had to be altered. Hover over it to see the warning message. Often, there was more than one potential position for an atom so we chose one. If it was a non-canonical residue, it will indicate what canonical residue has been chosen to replace it.
if a residue had to be altered. Hover over it to see the warning message. Often, there was more than one potential position for an atom so we chose one. If it was a non-canonical residue, it will indicate what canonical residue has been chosen to replace it.
By default, waters will be removed, but you can select individual waters to be added back. They will not appear in this list until you choose to make it visible.
Again, only add a water molecule back if you are certain that it is necessary. If you know that an individual water is needed, you can select it to be added First, allow waters to become visible by clicking the ![]() button on the top of the window. Change water from hidden to visible by un-clicking the box next to water in the Deleted Waters row.
button on the top of the window. Change water from hidden to visible by un-clicking the box next to water in the Deleted Waters row.

This will add waters to the list of deleted molecules on the right and water molecules will appear in the Structure Viewer as black dots. Find the correct water molecule that needs to be kept and select it. Then click ![]() .
.
Warning: Structures loaded with DNA or RNA will be in a conformation that is dependent on that interaction. Since our current version of CAD cannot model DNA or RNA, we recommend that you load a structure that was experimentally derived without DNA or RNA. Alternatively, you can do structure optimization with Relax in order to find a conformation of your apoprotein, but there is a strong probability that the “optimized” structure will be heavily biased by any binding partners that were present. Most proteins make conformational changes when binding a partner. Some can be very different than the apo-conformation. Particularly at the binding site, of course. Relax will only find the new conformation if there is minimal differences (under 2 Å rmsd change).
In the example above, positions 401 and 402 are the molecules NAP and AV5. These are the labels of two molecules found in the pdb file. The full name of the molecule can be found by viewing the pdb entry for the protein 5OU0. You can click these positions here. You can verify that they are chosen because they light up in the Structure Viewer. In order to add these molecules to the set of atoms that will be loaded into CAD, click the ![]() button above the Structure Viewer.
button above the Structure Viewer.
Once you have included all the molecules you want, click ![]() which is in the top right section of the center window.
which is in the top right section of the center window.
The structure will automatically begin to be processed in order for the final changes to be loaded into CAD. It will appear in the list of structures in the Structure Loader tab:

Once it is loaded, a ![]() will replace the
will replace the ![]() . Also, the structure will appear in the list of loaded structures in the left window.
. Also, the structure will appear in the list of loaded structures in the left window.
Now if you click the new folder with your structure, you should see your changes.
The Ligand molecule will be normal CPK coloring with the carbons colored the same as the protein, or whichever custom color that you choose. (Note that only carbons can be recolored.)
HOW TO RUN ACTIONS WHEN A SMALL MOLECULE IS PRESENT
There are a few things you should keep in mind while modeling structure with small molecules.
The likely worst case is that your small molecule will become ejected from it’s binding pocket. If this happens, there are a few ways to deal with it:
1.) It’s possible that you are modeling with a ligand in a pocket that is not a good fit, chemically or sterically. So it is normal behavior for the ligand to pop out of the pocket.
- If you intend to mutate residues around the ligand in order to optimize the affinity to the ligand, then you just need the ligand to stay put so you can sample mutations until you find the right mutations. While we normally recommend that you optimize your structure before running Design, in this case, you can take your non-optimized structure to use as the input structure for Design. Though, this can be a bit noisier, so more repeats of your Designs runs may be necessary to find the best mutations. Then post-Design Relaxes can verify that the mutations will optimize the structure without the ligand wanting to pop out of the pocket.
- If your ligand is known to bind the protein, but the pocket is not a good fit, it is probable that your need to find an alternate binding mode. Our software will not find that new mode. Bench will only make minor shifts in the ligand’s position. So, if the true binding mode requires more than a 1 angstrom shift, we will not find it. Use an alternative docking tool to find a better binding mode and then load that structure into Bench.
2.) It’s possible that the ligand is a good fit for the pocket where it is bound, but our software cannot properly define its chemistry so it is kicking it out of the pocket inappropriately.
- Not every small molecule will be well characterized by our software. We use Open Eye to analyze your ligands and create the parameters files that Rosetta can understand. In the next section, we describe how this is done so you can predict whether your ligand handling is your issue. If you suspect that is the case, email support@cyrusbio.com with a copy of the structure file and we can potentially find a fix. Your structure will be considered private data that we will not share.
- If you know you want to ligand present even if it our software wants to kick it out, there are a few tricks to persuade the ligand to stay put. However, always keep in mind that working with a ligand that the Rosetta score has determined not a good fit will skue results.
- Use Ligand Restraints
- Optimize structure with Prepare, rather than with rounds of Relax until achieving minimized score. Prepare is a constrained Relax that attempts to prevent rmsd shift over 1 rmsd. However, you should still verify that the ligand is near your desired position.
- You may run a large number of Relax repeats, then look at the output structures to find one that keeps your ligand position with the lowest score. This could be tedious if you run 100 repeats and it turns out the 30 best scoring structures move the ligand.
- You can also try Repack and/or Minimize, which are less aggressive that Relax, yet can improve the score.
All small molecule will be modeled with flexibility. When small molecules are initially loaded, a library of energetically-favorable conformations of each molecule is created. This library has up to 200 conformations of the small molecule. Most molecules will not have that many conformations because they don’t have enough rotatable bonds for that much variation. The Open Eye software creates the first conformer when it finds the most energetically favorable conformation. The second conformer will be the second best energetically, but must differ from the first by an rmsd cutoff. Remaining conformers are selected in the same way until no more conformations are possible or the 200th conformer is found.
You may want to increase the amount of modeling done for larger molecules. For example, we recommend running 50 repeats of Relax for a protein in order to find a conformation with lower energy. It is usually necessary to take the lowest energy Relax conformation and run an additional 50 repeats of Relax in order to full optimize the structure. This may be necessary multiple times in order to find the lowest possible score. More rounds of Relax are likely to be needed when many, large small-molecules are present because more sampling could be necessary to find the energetic minimum.
WHAT YOU NEED TO KNOW ABOUT LOADING A PROTEIN WITH NON-PROTEIN ATOMS
- You still need a pdb file
- You can load a file without a protein, but most Actions will not run (as you would expect)
- You need to decide which atoms to keep before you can perform Actions
- Not every molecule will work out
There are a few reasons why a molecule could fail to function well in CAD. Open Eye is incredible software for defining molecules, but they trained their software on known molecules with experimental data. This limits their accuracy to things with experimental data and things that can be readily extrapolated from that data. Hypothetical molecules run a higher risk of modeling incorrectly. This does not rule out errors involving well known small molecules. For example, heme is a known molecule that is difficult to model because iron is not well parameterized.
When a small molecule is loaded into CAD, we use Open Eye to assign the partial charges. The preferred method is called AM1-BCC. If this is not possible, we use the method called Gasteiger. If that is also not possible, we leave the molecule uncharged. Currently, the user will NOT get an error message when this occurs. We will soon add an error message so the user can decide if they want to continue with this non-ideal charge assignment. User will be told either that the small molecule parameters file is expected to be strongly-confident that it representative of the chemistry, reasonably-confident, or low-confidence (likely to have errors when modeling).
CAD will also create a library of conformations for every small molecule that you load, as described above. If it is unable to determine bond order or geometry, it will not be able to do so. In that case, the input conformation will be remain rigid during modeling.
For More Information on how small molecules are generated and modeled in Rosetta:
Generating Partial Charges:
Jakalian A, Jack DB, Bayly CI. Fast, efficient generation of high-quality atomic charges. AM1-BCC model: II. Parameterization and validation. J Comput Chem. 2002 Dec;23(16):1623-41.
Generating Conformers:
Takagi T, Amano M, Tomimoto M. Novel method for the evaluation of 3D conformation generators. J Chem Inf Model. 2009 Jun;49(6):1377-88.
Generating Rosetta Params steps 17-19:
Combs SA, Deluca SL, Deluca SH, Lemmon GH, Nannemann DP, Nguyen ED, Willis JR,Sheehan JH, Meiler J. Small-molecule ligand docking into comparative models with Rosetta. Nat Protoc. 2013;8(7):1277-98.
Comparing Rosetta Ligand performance:
Davis IW, Raha K, Head MS, Baker D. Blind docking of pharmaceutically relevant compounds using RosettaLigand. Protein Sci. 2009 Sep;18(9):1998-2002.
OVERVIEW
Loading a structure file into Bench is a two step process.
Step One: Select which components of the structure file you wish to be included.
Step Two: Bench processes the file contents to define all molecules in a way that allows Rosetta to understand the physics of everything present.
Importing a structure into Bench can be a very simple or complex process depending on what is present in your file. When only loading a protein, there is not much to consider, but the handling of non-protein molecules requires some potentially complex, but important steps.
The conformation and sequence preferences of a protein can change significantly when bound to another molecules. So, the ability to model proteins with their non-protein binding partners is hugely important. Rosetta is excellent at doing so, but it takes a great deal of work to create the files that will provide the physics-based descriptors for each ligand in order for Rosetta to model them properly. This is a cumbersome task in Rosetta. Unique to Cyrus Bench is the ability to automatically and reliably parameterize non-peptidic residues. The automated parameterization process integrates characterization tools from Rosetta with additional tools from the OpenEye small molecule toolkit. Open Eye is a leader in the field of small molecule cheminformatics and their toolkits are widely used by small molecule bioinformaticists.
LOADING A PROTEIN WITH SMALL MOLECULES
When you load a pdb file into CAD that contains non-protein elements, those elements can be processed and included in the system. Rosetta does not have an automated way to understand non-proteins, so they require expert handling. Rosetta already has extensive software for automated characterization of a protein. It can quickly load a pdb and create a profile that defines it’s chemistry. As complex as proteins are, they are all composed of the same 20 amino acids which have well characterized physics. The chemical space required to define non-peptidic molecules is much more expansive. Defining the physics of non-peptidic molecules requires analysis of the topology and subsequent determination of atom types, partial charges, and rotomeric preferences.
Important Note: We currently do not allow you to keep DNA, or RNA. We also limit the protein to the 20 canonical amino acids. We convert non-canonical amino acid residues to their closest canonical parent. We will eventually allow DNA, RNA, and non-canonical residues. These can be very important, but further code development is necessary to include proper modeling. Because Rosetta/Bench uses an implicit solvent water model, most/all crystallographic waters should typically be discarded. On occasion, a water serves a specific structural purpose, such as bridging interactions between a small molecule ligand and the protein. In that case, you may wish to include a very limited number of explicit waters.
HOW TO LOAD A STRUCTURE WITH SMALL MOLECULES PRESENT
You can load a structure that has a small molecule with the Structure Loader:
You can enter your PDB ID or load a file from your computer as usual.
In the next step, you will be asked which non-protein molecules from the file you wish to keep. By default, all protein, DNA, and RNA residues are kept and other atoms are shown as things you do NOT want included. However, you can add back or remove anything.
In the top of the tab window, there are 3 drop down buttons. In this example, the first reads ![]() . (The number will change depending on how many residues are present.) Clicking this button reveals a summary of what residues and molecules are visible in the Structure Viewer. Any changes you make in this box will change the appearance in the workspace, but don’t by themselves delete or undelete elements of the system. Two examples are shown below.
. (The number will change depending on how many residues are present.) Clicking this button reveals a summary of what residues and molecules are visible in the Structure Viewer. Any changes you make in this box will change the appearance in the workspace, but don’t by themselves delete or undelete elements of the system. Two examples are shown below.
Anything that has its box checkmarked will be visible, even if it is set to be deleted. Anything not checkmarked will not be visible, even if it is set to be included. In the first column, you can see that 5OU0 has 1 chain (A) and 1Z7J has 2 chains (A and B). Later in this article, you will be shown how to remove chains.
In the second column, the types of molecules selected are shown. You can see that the type of molecules currently selected is only the Protein molecules. Our default mode is to only keep the protein, DNA, and RNA. The second item in the list (NC Protein) refers to non-canonical proteins. We currently convert all NC Proteins to canonical. So, for example, if you have Selenocysteine on your protein, it will be converted to a Cysteine. The others are self explanatory, except for Ligand. This term is used for anything else.
The third column lists the things currently removed from the Structure Viewer.
As you add things to be included in the final structure load, things will become visible in the Viewer. That will be indicated in this window as well.
The next button, ![]() will allow you to alter which things you have currently selected.
will allow you to alter which things you have currently selected.
This can speed things up if you know you want to include everything. Or deselect everything if you know you only want to choose a few things manually. Or if you want to select all the things that have been automatically changed during import, meaning the non-canonical amino acids. And finally, you can select all the deleted items, typically the mono-atomic or ligand molecules. Again, DNA and RNA will not be allowed until the next software upgrade.
The third button on the top says ![]() by default, but can be switched to
by default, but can be switched to ![]() . This is helpful if a crystal includes multiple copies of the relevant protein or protein complex in the unit cell. When in
. This is helpful if a crystal includes multiple copies of the relevant protein or protein complex in the unit cell. When in ![]() mode, you can add or remove whole chains in the Structure Viewer. Select the elements you want to modify, then click
mode, you can add or remove whole chains in the Structure Viewer. Select the elements you want to modify, then click ![]() or
or ![]() .
.
The workspace, which appears below, presents a visual view of the molecules in the PDB file. Clicking on various elements of the structure, along with using the buttons above, provides the user with another way to add/delete included elements. Deleted elements of the system appear black.
Above the Structure Viewer is the sequence. Mono-atomic, water, and Ligand items appear after the protein. Any deleted elements are given the single letter x in the sequence. If you undelete any of these elements, it becomes z in the sequence or w for water. Any protein residues that are deleted will also be represented with an x.
Finally, the panel to the right of the Structure Viewer lists the individual items that are currently deleted or that have been altered.
They appear in order of their residue number. The next column will have ![]() if a residue had to be altered. Hover over it to see the warning message. Often, there was more than one potential position for an atom so we chose one. If it was a non-canonical residue, it will indicate what canonical residue has been chosen to replace it.
if a residue had to be altered. Hover over it to see the warning message. Often, there was more than one potential position for an atom so we chose one. If it was a non-canonical residue, it will indicate what canonical residue has been chosen to replace it.
By default, waters will be removed, but you can select individual waters to be added back. They will not appear in this list until you choose to make it visible.
Again, only add a water molecule back if you are certain that it is necessary. If you know that an individual water is needed, you can select it to be added First, allow waters to become visible by clicking the ![]() button on the top of the window. Change water from hidden to visible by un-clicking the box next to water in the Deleted Waters row.
button on the top of the window. Change water from hidden to visible by un-clicking the box next to water in the Deleted Waters row.
This will add waters to the list of deleted molecules on the right and water molecules will appear in the Structure Viewer as black dots. Find the correct water molecule that needs to be kept and select it. Then click ![]() .
.
Warning: Structures loaded with DNA or RNA will be in a conformation that is dependent on that interaction. Since our current version of CAD cannot model DNA or RNA, we recommend that you load a structure that was experimentally derived without DNA or RNA. Alternatively, you can do structure optimization with Relax in order to find a conformation of your apoprotein, but there is a strong probability that the “optimized” structure will be heavily biased by any binding partners that were present. Most proteins make conformational changes when binding a partner. Some can be very different than the apo-conformation. Particularly at the binding site, of course. Relax will only find the new conformation if there is minimal differences (under 2 Å rmsd change).
In the example above, positions 401 and 402 are the molecules NAP and AV5. These are the labels of two molecules found in the pdb file. The full name of the molecule can be found by viewing the pdb entry for the protein 5OU0. You can click these positions here. You can verify that they are chosen because they light up in the Structure Viewer. In order to add these molecules to the set of atoms that will be loaded into CAD, click the ![]() button above the Structure Viewer.
button above the Structure Viewer.
Once you have included all the molecules you want, click ![]() which is in the top right section of the center window.
which is in the top right section of the center window.
The structure will automatically begin to be processed in order for the final changes to be loaded into CAD. It will appear in the list of structures in the Structure Loader tab:
Once it is loaded, a ![]() will replace the
will replace the ![]() . Also, the structure will appear in the list of loaded structures in the left window.
. Also, the structure will appear in the list of loaded structures in the left window.
Now if you click the new folder with your structure, you should see your changes.
The Ligand molecule will be normal CPK coloring with the carbons colored the same as the protein, or whichever custom color that you choose. (Note that only carbons can be recolored.)
HOW TO RUN ACTIONS WHEN A SMALL MOLECULE IS PRESENT
There are a few things you should keep in mind while modeling structure with small molecules.
The likely worst case is that your small molecule will become ejected from it’s binding pocket. If this happens, there are a few ways to deal with it:
1.) It’s possible that you are modeling with a ligand in a pocket that is not a good fit, chemically or sterically. So it is normal behavior for the ligand to pop out of the pocket.
- If you intend to mutate residues around the ligand in order to optimize the affinity to the ligand, then you just need the ligand to stay put so you can sample mutations until you find the right mutations. While we normally recommend that you optimize your structure before running Design, in this case, you can take your non-optimized structure to use as the input structure for Design. Though, this can be a bit noisier, so more repeats of your Designs runs may be necessary to find the best mutations. Then post-Design Relaxes can verify that the mutations will optimize the structure without the ligand wanting to pop out of the pocket.
- If your ligand is known to bind the protein, but the pocket is not a good fit, it is probable that your need to find an alternate binding mode. Our software will not find that new mode. Bench will only make minor shifts in the ligand’s position. So, if the true binding mode requires more than a 1 angstrom shift, we will not find it. Use an alternative docking tool to find a better binding mode and then load that structure into Bench.
2.) It’s possible that the ligand is a good fit for the pocket where it is bound, but our software cannot properly define its chemistry so it is kicking it out of the pocket inappropriately.
- Not every small molecule will be well characterized by our software. We use Open Eye to analyze your ligands and create the parameters files that Rosetta can understand. In the next section, we describe how this is done so you can predict whether your ligand handling is your issue. If you suspect that is the case, email support@cyrusbio.com with a copy of the structure file and we can potentially find a fix. Your structure will be considered private data that we will not share.
- If you know you want to ligand present even if it our software wants to kick it out, there are a few tricks to persuade the ligand to stay put. However, always keep in mind that working with a ligand that the Rosetta score has determined not a good fit will skue results.
- Use Ligand Restraints
- Optimize structure with Prepare, rather than with rounds of Relax until achieving minimized score. Prepare is a constrained Relax that attempts to prevent rmsd shift over 1 rmsd. However, you should still verify that the ligand is near your desired position.
- You may run a large number of Relax repeats, then look at the output structures to find one that keeps your ligand position with the lowest score. This could be tedious if you run 100 repeats and it turns out the 30 best scoring structures move the ligand.
- You can also try Repack and/or Minimize, which are less aggressive that Relax, yet can improve the score.
All small molecule will be modeled with flexibility. When small molecules are initially loaded, a library of energetically-favorable conformations of each molecule is created. This library has up to 200 conformations of the small molecule. Most molecules will not have that many conformations because they don’t have enough rotatable bonds for that much variation. The Open Eye software creates the first conformer when it finds the most energetically favorable conformation. The second conformer will be the second best energetically, but must differ from the first by an rmsd cutoff. Remaining conformers are selected in the same way until no more conformations are possible or the 200th conformer is found.
You may want to increase the amount of modeling done for larger molecules. For example, we recommend running 50 repeats of Relax for a protein in order to find a conformation with lower energy. It is usually necessary to take the lowest energy Relax conformation and run an additional 50 repeats of Relax in order to full optimize the structure. This may be necessary multiple times in order to find the lowest possible score. More rounds of Relax are likely to be needed when many, large small-molecules are present because more sampling could be necessary to find the energetic minimum.
WHAT YOU NEED TO KNOW ABOUT LOADING A PROTEIN WITH NON-PROTEIN ATOMS
- You still need a pdb file
- You can load a file without a protein, but most Actions will not run (as you would expect)
- You need to decide which atoms to keep before you can perform Actions
- Not every molecule will work out
There are a few reasons why a molecule could fail to function well in CAD. Open Eye is incredible software for defining molecules, but they trained their software on known molecules with experimental data. This limits their accuracy to things with experimental data and things that can be readily extrapolated from that data. Hypothetical molecules run a higher risk of modeling incorrectly. This does not rule out errors involving well known small molecules. For example, heme is a known molecule that is difficult to model because iron is not well parameterized.
When a small molecule is loaded into CAD, we use Open Eye to assign the partial charges. The preferred method is called AM1-BCC. If this is not possible, we use the method called Gasteiger. If that is also not possible, we leave the molecule uncharged. Currently, the user will NOT get an error message when this occurs. We will soon add an error message so the user can decide if they want to continue with this non-ideal charge assignment. User will be told either that the small molecule parameters file is expected to be strongly-confident that it representative of the chemistry, reasonably-confident, or low-confidence (likely to have errors when modeling).
CAD will also create a library of conformations for every small molecule that you load, as described above. If it is unable to determine bond order or geometry, it will not be able to do so. In that case, the input conformation will be remain rigid during modeling.
For More Information on how small molecules are generated and modeled in Rosetta:
Generating Partial Charges:
Jakalian A, Jack DB, Bayly CI. Fast, efficient generation of high-quality atomic charges. AM1-BCC model: II. Parameterization and validation. J Comput Chem. 2002 Dec;23(16):1623-41.
Generating Conformers:
Takagi T, Amano M, Tomimoto M. Novel method for the evaluation of 3D conformation generators. J Chem Inf Model. 2009 Jun;49(6):1377-88.
Generating Rosetta Params steps 17-19:
Combs SA, Deluca SL, Deluca SH, Lemmon GH, Nannemann DP, Nguyen ED, Willis JR,Sheehan JH, Meiler J. Small-molecule ligand docking into comparative models with Rosetta. Nat Protoc. 2013;8(7):1277-98.
Comparing Rosetta Ligand performance:
Davis IW, Raha K, Head MS, Baker D. Blind docking of pharmaceutically relevant compounds using RosettaLigand. Protein Sci. 2009 Sep;18(9):1998-2002.